[ed. note: Michael Jorrin, who I like to call Doc Gumshoe, is a longtime medical writer (not a doctor) who writes about non-financial health and medical issues for us a couple times a month (past submissions can be found on his author page here). Like all of our authors, he chooses his own topics and his words and opinions are his alone. Enjoy!]
Okay, I needed a catchy title for this piece, but you already know the answer to that pretend-question. There are no archvillains, except maybe in grade D horror movies, and certainly not in disease management. But the temptation to pin that label on something such as inflammation, and then pick a “cure” that deals with that dire threat, is very substantial. It’s reassuring to fix in our minds that the chief cause of lung cancer is cigarette smoking, and if we avoid that particular archvillain, we won’t get lung cancer. Or that the chief cause of diabetes is drinking sugary soda pop. Or, as was doctrine for quite some time, that the chief cause of heart disease is feasting on red meat, eggs, and butter. Targeting an archvillain leads to an easy and quick solution, and minimizes the tedious and often confusing investigation into complex mechanisms and interrelations between external causes and internal physiologic processes.
Ascribing unitary causes to physical phenomena works quite well in the physical sciences, even when a secondary cause might also participate in the effect. In particular, it makes the investigation more transparent. Here’s an observed phenomenon. We postulate one specific direct cause. We then test it – does the phenomenon occur every single time when we apply that one specific direct cause? If it does, we’ve nailed it. If it only happens some of the time, or if it happens in the absence of that specific cause, then something else must be the cause. Our hypothesis has been proved false. Then we have to keep going until we find the precise action that always produces that effect. And then we dig until we figure out exactly why that action produces that effect. That works in the physical sciences, but not so well in the biological sciences.
In living organisms, a single cause can have multiple effects, and in some cases, can have effects that tend in contrary directions. For example, we know that diabetes can lead to high levels of glucose in the bloodstream, and those high levels of glucose can lead to serious consequences, including blindness, limb amputations, and heart disease. But we absolutely and unquestionably require glucose for life, and it has to be transported throughout the body in the bloodstream. So the issue is not whether glucose in the bloodstream is the archvillain. The question is how much is too much, and what can be done to avoid getting to that “too much” marker.
We also know that cancer occurs as a result of errors in the transcription of genetic material in stem cell division. Every time a stem cell divides, there is a chance that it will make a mistake, such that the “offspring” cells are not exact copies of the “parent” cell. These inaccurate cell divisions are essentially random mutations, and the great majority of these mutations will be meaningless – the new cells will simply die or fail to replicate. In a few cases, the mutations will be beneficial, conferring some evolutionary advantage, and these mutations may become part of the species genome. And in some cases, the mutation will lead to cancerous cells, which continue to replicate and increase in number. So, we cannot say that mutations in stem cell division are the archvillains that cause cancer, and that if we could prevent those mutations, cancer would be prevented. It is also those mutations that drive evolution, and without them we – and most of the rest of living beings – would not have come into existence.
The same principle applies to inflammation. Although we are increasingly aware of the role of inflammation in a number of different pathologies, we also know that inflammation is an essential and integral part of the healing process. This knowledge is far from new. Hippocrates, in the 5th century BC, used the term edema in his description of the healing process, and Aulus Celsus, who lived just around the beginning of the first century AD, described the four signs of inflammation as rubor (redness), tumor (swelling), calor (heat), and dolor (pain). Inflammatio means “setting on fire,” and everyone one who has sustained just about any kind of injury knows what that feels like.
Here’s a look at the inflammatory process. You sustain an injury, not severe enough to send you to the emergency room, but definitely painful – let’s say you scraped your leg while climbing over a stone wall. The scrape only slightly broke the skin, and there was just a bit of bleeding, but the impact was painful. This event triggers a sequence of events, beginning with a considerable increase in local blood flow. The capillaries become more permeable, resulting in the build-up of fluid in the space in and around muscle and nerve fibers. A large number of cells of many different types are summoned to the area, and these immediately go to work to clean up the mess. Damaged tissue is devoured and carried off by macrophages and neutrophils, and the process of replacing dead cells with new cells begins. The entire area of the injury is walled off, so as to prevent the spread of any invading pathogens. And, yes, even though your skin was only slightly broken, pathogens certainly did enter. That’s because your skin (no matter how scrupulous you are about personal hygiene) is colonized by immense numbers of microbes of various kinds, especially staphylococci, which are primed to attack your cells. The inflammatory process is highly efficient at preventing the spread of staph infections that invade in that way. Once the infection has been contained by this walling-off process, your own immune system will wage war on the invading pathogens, and in most cases, your immune system will achieve victory in this war.
One could say that the reason the inflammatory process results in redness, swelling, heat, and pain, is that inflammation confines and concentrates the injury to a relatively small area, like waging an all-out war on a single small battlefield.
And, naturally, as in any war, there is collateral damage. If the inflammation occurs as a result of a bruise or a minor skin infection, the collateral damage will be minor. But if, on the other hand, inflammation occurs as a response to some type of internal disruption or as a reaction to certain types of stimuli, the collateral damage can be considerable. And, in some cases, inflammation seems to occur spontaneously, not in response to injury or a specific stimulus. In those cases, the inflammatory process in itself constitutes the disease.
Mediators of inflammation
The very word “mediators” in that context is a bit confusing, at least to the non-medical reader. Most people think of mediators as the folks who try to resolve differences between two warring parties, like the opposite sides of a lawsuit or labor unions versus management. In medical terms, the mediators are the agents that trigger the response, period. There is no give-and-take.
The numbers of inflammatory mediators are legion. They can be relatively simple molecules or living cells. Some of the ones whose names most people will recognize include histamines, serotonin, prostaglandins, bradykinins, a number of cells carried in the bloodstream such as macrophages and neutrophils, B-cells, T-cells, lymphokines, caspases, and many, many others. A number of factors also stimulate generation of granulocytes and monocytes by the bone marrow; these include tumor necrosis factor (TNF), which is one of the drivers of rheumatoid arthritis (RA).
A large category of inflammatory mediators are classified as cytokines, although by no means are all cytokines pro-inflammatory. I attended a meeting of the American College of Rheumatology where a scientist attempted to present an overview of the cytokines. Rheumatology, by the way, is the branch of medicine that studies inflammatory diseases; “rheum” being the old-timey word for the wet discharge from the eyes or nose, and also for the substance that accumulates in areas affected by inflammation. Thus “rheumatism” was the word for what is now called arthritis. Rheumatologists are the medical specialists that also treat other diseases of the immune system, such as inflammatory bowel disease (IBD), Crohn’s disease, psoriasis, and ankylosing spondylitis.
The presenter began with a simple slide that mostly showed the interleukins, a well-studied group of cytokines, some of which are clearly pro-inflammatory and some, quite the contrary, are anti-inflammatory. There were about twenty on that slide. He then went on to show a slide with about one hundred cytokines on it, in different groups, in boxes connected by arrows. Then he put a slide on the screen that had so many cytokines on it that no one could possibly read one single name. He admitted that it was possible and even likely that many of the cytokines listed on the slide were identical. For example, a scientist in a lab in Cambridge might have identified this one, and a scientist in Palo Alto might have identified that one, and they were the same cytokine, but no one had spotted it. The audience erupted in laughter.
I put in that little story not to make the Gumshoe Tribe erupt in laughter, but to illustrate how the research goes. Scientists see something going on, and they investigate all the different particles – individual molecules or cells – and try to determine what roles they are playing in what’s going on. It’s easy to see what macrophages are doing. They are huge (comparatively) and one can actually watch them in action. Besides cleaning up the mess that occurs when pathogens invade and create an infection, macrophages are capable of pumping out many different small molecules that also contribute to the inflammatory process.
The small molecules such as cytokines are a different matter. Scientists detect the presence of such a molecule in association with an inflammatory process, and then they try to figure out what its role might be, if any. Cytokines do not engulf and gobble up bacteria or cells. Instead, they may interact chemically with receptors on the surface of these bodies, or they may pass through openings in their coating. These actions depend on the precise shape of the molecules and on their binding properties. Sometimes a molecule fits neatly into receptors on the surface of a cell, thereby inactivating the receptor. The activity of some of these molecules may be conceptually simple, but detecting and understanding that activity is far from simple.
Consequences of inflammation beyond the known autoimmune diseases
The evidence for this has been around for about 30 years. Before that, arthritis, at least, was thought to be a relatively benign disease. Old people with arthritis could hobble around with canes, or in the most severe cases, could get around on wheelchairs, but arthritis, whether osteoarthritis or autoimmune rheumatoid arthritis, didn’t kill people.
That notion began to be exploded in 1984 when a rheumatologist named Theodore Pincus caused a considerable stir at an ACR meeting when he reported that in a group of 75 patients with RA who had been tracked for 9 years, the mortality rate was about the same as in patients with heart disease blocking three coronary arteries – i.e., really severe disease. By the mid 1990s, there were lots of data showing that, in fact, rheumatoid arthritis did kill people – or, at least, that patients with rheumatoid arthritis were more than twice as likely to die as people of the same age in the general population.
But how on earth did this disease, which apparently affected only the joints, result in fatalities? It had been observed that persons with RA tended to have higher incidences of cardiovascular disease than those without RA, and it was conjectured that a possible reason for this was that RA patients were much more limited in terms of physical activity. In the view of some, lack of exercise was the culprit that was consigning RA patients to a premature death.
That line of reasoning appears to be sound, up to a point, in that physical activity certainly does contribute to cardiovascular health, and inactivity does the opposite. But several kinds of data began to appear about 20 years ago that suggested a different mechanism. Some of the data was statistical and some was the result of close scientific observation.
The mid-1990s, you will perhaps remember, were an era in which elevated cholesterol had been confirmed, in the view of most cardiologists, as the essential cause of coronary artery disease. It had been established that atherosclerosis consisted of cholesterol deposits in the arteries, and the recent 4S trial had conclusively shown that in patients with heart disease, statin treatment greatly reduced the incidence of significant cardiac events such as heart attacks and obstruction of coronary arteries requiring revascularization. In other words, problem solved: cholesterol is the culprit.
"reveal" emails? If not,
just click here...
Some data got in the way of this unitary explanation. One was that a certain number of individuals who had “normal” cholesterol levels nonetheless experienced the same kind of cardiac events. Paul Ridker, a cardiologist at Brigham and Women’s hospital and Harvard Medical School, found that these persons, who did not have cholesterol at levels that had been associated with heart disease, did have elevated levels of C-reactive protein (CRP), which for more than 80 years has been known to be associated with generalized inflammation.
At about the same time, another Brigham and Women’s/ Harvard cardiologist, Peter Libby, learned that cholesterol didn’t just swim around in the bloodstream. It actually worked its way into the arterial wall. This appeared to constitute a kind of insult to the arterial wall and provoked an inflammatory response, which in turn resulted in the formation of blood clots. It was these blood clots that, at least in some cases, blocked coronary arteries, causing heart attacks, and also blocked cerebral arteries, causing strokes. Peter Libby coined the term “vulnerable plaque” for plaque affected by inflammation that was prone to clot formation.
(Now, lest the above information confirm the views of those who claim that it’s not cholesterol but inflammation that is the archvillain, let me insert a modest demurral, viz, lots of factors besides inflammation can cause the formation of blood clots. Blood tends to clot all on its own, and conditions such as atrial fibrillation, in which blood pools in the heart antechambers, or venous thrombosis, favor the formation of blood clots.)
Paul Ridker followed up his discovery about CRP with a study in which it was shown that treatment with statins not only lowered cholesterol levels, but also lowered levels of this inflammation marker. And in 2008, Ridker presented the results of the JUPITER trial at the New Orleans meeting of the American Heart Association. (Ridker P et al. New Engl J Med 2008;359:2195-2207) This large trial (17,802 subjects) compared two cohorts of persons, all of whom had normal cholesterol levels. One group of 8,901 subjects received 20 mg. of rosuvastatin daily, and the other, also 8,901, got the placebo. The primary endpoint, was incidence of signal cardiac events consisting of nonfatal myocardial infarction, nonfatal stroke, unstable angina, or death from cardiovascular causes. Subjects receiving rosuvastatin experienced 142 such events, while those on placebo experienced 251 events. Although the reduction was small in terms of absolute risk – about 1.2% – it was considered highly significant, both statistically and in terms of implications for treatment. As a result of these results, the trial was stopped after a bit less than two years because the sponsors considered it unethical to continue a large cohort of patients on placebo when significant benefit had been demonstrated in the treatment arm.
The subjects in the JUPITER trial had baseline LDL-cholesterol levels of 108 mg/dL and CRP levels of 4.2/4.3 mg/L. Those LDL-C levels are considered desirable in patients with no established cardiac risk factors. CRP levels > 4.0 mg/L are now considered elevated and associated with significant risk.
The JUPITER trial cannot be said definitely to demonstrate that lowering CRP was the determining factor in reducing the numbers of signal cardiac events. Treatment with rosuvastatin not only reduced CRP from the baseline level to about 1.8 mg/L, but also lowered the LDL-C levels from a pretreatment 108 mg/dL to 55 mg/dL, so the benefit may have in part been due to the LDL-C reduction. But the reduction in that marker of inflammation was certainly an eye-opener. At the AHA meeting, Steven Nissen of the Cleveland Clinic was quoted as follows: “…if a patient comes to me with normal LDL-cholesterol levels, I tell him to keep doing what he’s doing and to go about his business. Now, what happens when that same patient arrives in my office and I know his CRP is elevated? I know that treating him with intensive statins therapy, despite what the guidelines state, is going to cut his risk of cardiovascular morbidity and mortality in half.”
In further analysis of the JUPITER trial results, Ridker came to the conclusion that elevated CRP levels signal more heart disease risk than do elevated LDL-C levels, although the highest risk is in patients in whom both of those are elevated. CRP levels below 1 mg/L are related to low risk, between 1 and 3 mg/L to medium risk, and higher than 3 mg/L to higher risk. The chart below traces the relationship between LDL-C and CRP levels and cardiac risk over an 8-year period.
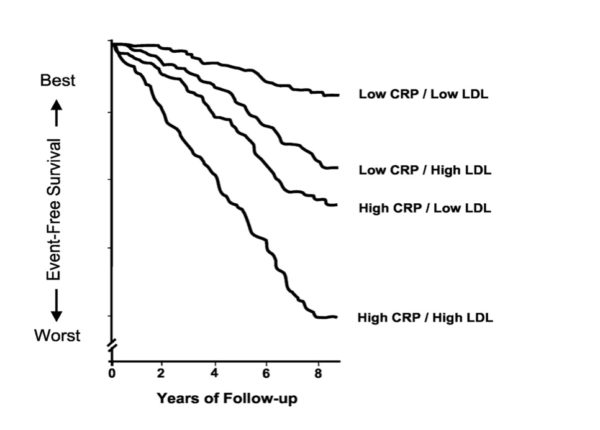
Another graphic presents a similar conclusion, that the highest level of risk is in those persons with elevated levels both of LDL-C and CRP.

The LDL-C levels considered as low in these graphics are based on the guidelines promulgated by the National Cholesterol Educational Program (NCEP) Adult Treatment Panel III in 2001, which used 130 mg/dL as the cutpoint for borderline high LDL-C. More recently, many clinicians have argued for a target LDL-C reading of about 100 mg/dL in individuals with no specific cardiac risk factors, and below 70 mg/dL in persons with one or more risk factors. Thus, the data above could be criticized as minimizing the risk presented by elevated levels of LDL-C.
Although the data as presented strongly suggests that inflammation is a greater risk factor for heart disease than elevated cholesterol, it is far from presenting satisfactory vindication for the supporters of the view that inflammation is the whole story and cholesterol is a mere ruse cooked up by the pharmaceutical industry as a way to push their products. After all, the JUPITER trial demonstrated that a statin lowered both CRP and LDL-C, and in so doing conferred significant reductions in heart disease risk in a cohort of persons who, according to the standards at the time, were not considered to have any heart disease risk at all.
Managing heart disease risk in patients with RA and other inflammatory diseases
The JUPITER trial, and several others, offered strong evidence that treating RA patients with statins, even when their LDL-C levels would not have – at least according to guidelines – led to such treatment, significantly reduced their risk of cardiovascular events. The benefits of other forms of treatment are somewhat more questionable.
The usual progression of treatment for persons affected by arthritis is from NSAIDs (non-steroidal anti-inflammatory drugs) such as ibuprofen, naproxen and others, through DMARDs (disease-modifying anti-rheumatic drugs) such as methotrexate (MTX), and ultimately biologics, such as tumor-necrosis factor inhibitors (TNFi’s) and agents that affect one of the pro-inflammatory cytokines. These individuals are also sometimes treated with glucocorticoids, although not usually on a long-term basis.
It is in evaluating the effects of these forms of treatment that the vista gets murky. For many individuals affected by arthritic joint pain, the priority is pain management. Quite reasonably, they want to be able to do whatever it is they need to do, or enjoy doing, without the pain that accompanies movement. It is common for persons with arthritic pain to go for a considerable time without consulting a physician. Joints hurt? Pick up a bottle of some kind of pain killer from the drugstore.
The NSAIDs do mitigate inflammation, and to the degree to which they make it easier for the individual to remain reasonably active, they may slow the progression of factors that contribute to cardiovascular disease. But some NSAIDs do increase the risk for both MIs and strokes. In June of 2015 the FDA beefed up the warning label on prescription NSAIDs, and various professional groups have urged physicians to prescribe NSAIDs at the lowest possible dose and for the shortest possible time. This does not mean that if you wrench your shoulder or sprain your ankle you should stay away from taking an NSAID; what it means is that regular high doses of NSAIDs for patients with chronic pain is not a good idea.
Similarly, there is evidence that prednisone is linked with higher cardiovascular risk. Again, that does not translate into “never prednisone,” just that a steroid as regular treatment for a chronic inflammatory ailment is bad medicine.
The methotrexate story is contradictory. Some evidence supports the view that MTX reduces cardiovascular events in RA patients by around 20% (Micha R et al. Am J Cardiol 108: 1362–1370). Other evidence points in the opposite direction. An analysis of the CORRONA registry, including data on more than 10,000 patients with RA, found that treatment with MTX in those patients did nothing to reduce their risk of cardiac events. (Greenberg JD et al. Ann Rheum Dis; 2011;70:576)
However, the same analysis found a considerable benefit in patients who were given TNF inhibitors. The incidence rate for composite cardiovascular events in patients who used TNFi’s was 2.93/1,000 patient-years of exposure, compared with 6.73/1,000 patient-years for methotrexate and 7.51 for the reference group of patients who used DMARDs, a 61% reduction in relative risk. Again, part of this benefit may be due to the increased capacity for activity and movement in the patients taking TNFi’s. Some observers attribute the benefit to specific anti-inflammatory effects in the vascular system, such as stabilization of arterial plaques and improvement in vasodilation, which facilitates blood flow.
Similar effects have been observed with biologics used in treating psoriatic arthritis, inflammatory bowel disease, Crohn’s disease, and others.
The role of inflammation in other medical conditions
Inflammation occurs in other parts of the body than those in which the diseases discussed above manifest themselves, i.e., the skin, the joints, the intestines, or the heart. Inflammation in the pancreas may contribute to diabetes, and inflammation in the brain may be a factor in Alzheimer’s disease. In both cases, the inflammation appears to be purely local. There is no good evidence that the rheumatic diseases are causally linked with either diabetes or Alzheimer’s disease.
Some observers have made the case that persons with RA have a higher incidence of diabetes. The weight of the numbers does not support that conclusion. Simply put, the percentage of RA patients with type 2 diabetes (T2DM) is roughly similar to the percentage of people in the general population with T2DM. In other words, lots of people with RA and other rheumatic diseases have T2DM, but so do lots of people who do not have those diseases.
But this does not mean that inflammation is not a factor. The question is whether inflammation in the pancreas is the cause of T2DM, or whether stress on the system of insulin receptors resulting in greater demand on the pancreas is what triggers the inflammatory response. On the other hand, type 1 diabetes is recognized as an autoimmune disease; inflammation by definition is part of the pathology.
It has been suggested by some that glucose on its own is proinflammatory. This is more than a bit paradoxical. After all, we live on glucose; it is our principal source of energy. According to this doctrine, it’s okay to consume foods that do not contain sugar or simple carbohydrates, such as whole grains. We can then convert those into glucose and continue on our merry way. But sugars and refined grains are verboten.
A prominent spokesman for this point of view is Mark Hyman, MD, founder of the UltraWellness Medical Center and director of the Cleveland Clinic Center for Functional Medicine. A book he co-wrote, The Daniel Plan, was a New York Times best-seller in 2013. His formula for curbing inflammation includes strict avoidance of sugar, caffeine, beans, all dairy foods, all foods containing gluten, and all processed foods. Instead, he proposes a variety of supplements including fish oil, probiotics, and several vitamins. He strongly favors turmeric, which contains curcumin.
We’ve discussed turmeric/curcumin in these posts before. (See “Somewhere Between ‘The Next Aspirin’ and ‘An Ingredient in Curry'”) Curcumin certainly does possess anti-inflammatory properties. The problem is that we cannot absorb enough of it in its natural form – i.e., from turmeric – to have much effect.
Hyman may be an effective physician in terms of managing the treatment of individual patients one-on-one. But my skepticism index regarding his one-size-fits-all proposal for achieving health by curbing inflammation through a strict diet based on supplements is as high as could be.
Hyman, by the way, is on the record as opposing the measles-mumps-rubella vaccine on the grounds that it leads to autism. Need I say more?
My own conclusion on this subject is, how shall I put it? – inconclusive. There’s no doubt that inflammation is an important factor in many diseases. But at the same time, it’s an immune response, and we absolutely require those immune responses to survive. Immune responses are what kill newly-formed cancer cells every day of our lives. Cancer researchers are currently investigating ways to harness immune mechanisms for cancer treatment. And, of course, without inflammation, healing would not take place.
But it is certainly possible that mechanisms that carefully dial down the immune reaction leading to inflammation may be identified and employed to reduce the impact of several diseases. The biologic agents used to treat RA essentially do just that. The simple solutions – diets and supplements – are not supported by any evidence. Too bad!
Doc enjoyed reading your article as I’ve RA – three years – and use Methotrexate and receive Humira needle once a fortnight.
My question is both of these really frighten me because as they can be damaging to your body over time, though they help keep RA under control.
Are there any other alternatives available in the market?
One possible solution for which there is no research ( because no rx company can profit and the government s not able to muster the $ for) is to look at food sensitivities. Beginning with research done by Peter D’Adamo’s father and first published in Eat Right 4 Your Type diet book, it was observed that certain people did better with less or more proteins, dairy and simple carbohydrates. An IGG test can help indicate a direction to pursue for an individual to see if making dietary changes will affect symptoms like pain and ongoing deterioration of joints. As a health coach and former member of an Institutional Review Board at a medical school for 22 years, I recognize both the value of the gold standard of studies and its limitations. Be brave, look past what is standard protocol and maybe you will find the evidence you need to improve your health. You can always go back to the drugs and if you get off be sure to ask for help in that process from your medical professionals so that the rebound doesn’t obscure any potential benefit from dietary or other modifications. I wonder if you looked at food sensitivity and began the change while on current meds if that might be useful. In my life I have found that physicians who practice Functional Medicine are more open to working with individual choices. Best wishes. Susan Levy CHES
Thanks Susan
Greetings Williamstown! (By the way, I lived there for a couple of years when I was a kid.) The MTX – Humira combo therapy is pretty standard & usually effective. MTX does have respiratory risks, as you doubtless have been warned. There are other biologics effective against RA than Humira, but all do have some adverse effects. The real question is, what’s the risk of RA progression & the risk that your RA will have other consequences. There is debate in the rheumatology community whether after a certain period in which RA is well-controlled, not only from the standpoint of symptoms, but also the markers of RA, patients may be able to be weaned off drugs & observed to see whether symptoms return. Some patients seem to be able to do well at least for a considerable period on “drug holidays.” Good luck!
Thank you appreciated
On the impact of NSAIDs, does use of a topical application (vis Voltaren) on source of inflammation/ pain mitigate the body-wide impact of a pill as medication (e.g. Aleve) ?
If using Voltaren or some other topical analgesic means you can reduce or omit the oral NSAID, that would “mitigate” the impact. But it depends on what you’re treating. If you have RA, quelling the symptom does not stop the progress of the disease, which entails the erosion of the joints. Remember, there are huge numbers of potential mediators of inflammation, and NSAIDs only target one type – prostaglandins. That said, I personally use Voltaren on my poor beat-up thumbs & rarely take any oral NSAIDs.
Very interesting and well-written article. But you need to update your research on vaccines in the light of CDC whistleblower Dr William Thompson coming out and admitting that data was concealed by the CDC linking the MMR vaccine to autism, especially among African American boys.
Not true: http://www.snopes.com/medical/disease/cdcwhistleblower.asp
Snopes, along with Glenn Kessler of the Washington Compost, is happy to carry water for a source with nearest/largest paycheck or anyone from the Democratic party. If there is a route straight to weasel they will take it. They do will contortions and torture data nearly as badly as Phil J0nes, Michael “hockey schtick” Mann and the rest of the watermelons of the climate cabal. Wealth redistribution the aim, climate change the game.
Speaking of which, personally, I can’t wait to see them fabricate an answer to Pat Franks devastating expose showing a systemic error in the calculation of the the climate models such that they don’t actually reflect reality….at all. LOL
https://wattsupwiththat.com/2016/11/22/the-needle-in-the-haystack-pat-franks-devastating-expose-of-climate-model-error/
The same models that are the primary source of the claims CO2 is responsible for global warming, er…climate change..er global climate disruption er…let just use their latin term, TheClimatisWhatWeSayitis.
And about that paper released just before the Paris get together that tried to show the pause didn’t happen. Opps! That’s gotta leave a mark.
http://www.climatedepot.com/2017/02/04/noaa-whistleblower-reveals-pausebuster-scandal/
Have you looked into Anatabine? From what I’ve heard it”s been shown to reduce inflammation with no side effects.
Ginger, Tumeric, Bromelain and Omega 3 are antiinflamatories. There is a study ongoing at Vanderbilt University testing the effects of Omega 3 regarding prevention of colon polyps.
Don’t know much about anatabine, other than what I’ve learned on-line which you likely know already. A study that reported benefit consisted of an on-line survey of a couple of hundred people who were already anatabine users; most said it helped. Compared to what? My guess is that they were converts already. Other studies have reported no benefit at doses 10 times higher than the highest commercially available preparation. My skeptical antennae are up.
Good research. Thanks for the work. Cytokines are Complicated by Design just as the blood clotting Cascade is intrinsically complicated. There are various feedback mechanisms that when they go awry by one mutation results in damage to the whole system and improper function. The cytokine diagram you mentioned demonstrates the complexity that is not yet elucidated, but what I do know is that some are pro-inflammatory and some are anti-inflammatory and they have various inter regulators. Too little inflammation can be just as big or worse of a problem then too much inflammation. I do think it is at the root of multiple illnesses in The Human Condition. Regarding sugar being the cause of all of human woes, I would say it’s too many calories.A new focus to cancer treatment is discussing the importance of weight reduction because of the pro-inflammatory aspects of obesity.
I quit reading when I read the ludicrous statement that we all came to being thru evolution. This theory has long been known to be false.
“This theory has long been known to be false.” You’re joking, right?
Longevity, mitochondrial rejuvenation, cellular renewal, losing weight and reduction of plaques in the brain due to or causation of Parkinson’s, dementia etc. Yes all this is all yours and the pill is FREE….How’s that for a teaser. The pill is actually called by its non medical name….. hard work. Specifically, caloric restriction and there a endless methods. Some supplements enhance the effect of the above benefits known as caloric restrictive autophagy such as…drum roll please…coffee and tea. Start googling autophgy, caloric restriction, intermittent fasting. Gain the knowledge and….well….begin the work (the only work that works). By the way, too much of the lack of inflammation due to anti inflammatory supplements can inhibit muscular synthesis but not by way of regular foods that contain them. Sometimes the body knows what it’s doing….if we let it.
Inflammation is the response. There’s something that causes that response in the first place. Find out what it is and the immune response doesn’t happen, be it physical injury, allergy, infection, or response of any sort. The mechanism of NSAIDs for example, prevents the response but does not heal the cause. When it comes to acute problems, especially physical trauma, Western Medicine is hard to beat. But not when it comes to chronic issues. Cancer treatments, arthritis treatments, they are all treatments of the symptoms and the immune response. Very few of them address the cause. Think of the word ‘treat-ment’, the etymology literally comes from ‘to manage the mind’. It is simply sick care, to make patients feel better, and has little to do with health or healing.There’s a million cures for everything you can imagine, just not in the form of an OTC product or a band aid.
I’d like to know if Doc Gumshoe has any info on
Chelation Therapy for unclogging arteries and
removing heavy metals .
“It has been suggested by some that glucose on its own is proinflammatory. ” Your discussion presumes ingestion of sugars may be bad for inflammation, and that might be the case.
However, hasn’t prolotherapy for decades used injection of dextrose to stimulate cell division of connective tissue through producing an inflammation response, which results in strengthening connective tissue and stabilization of joints? Modern prolotherapy uses stem cells. Unfortunately, there are fewer skilled prolotherapists than there are doctors giving single to a few injections and calling it “prolotherapy.” See for an introduction. The site archives a lot of literature, most of it peer-reviewed. I’ve used their services for over a decade to successfully heal various joint injuries, a couple that diagnosed as requiring surgery had I unquestioningly bought into the advice of surgeons. The folks at Caring Medical are legit.
Doc, I did enjoy reading the article and the study results as related to how prescription drugs can help resolve this or that issue. Your explanations are great. I often wonder though, if all that is studied is how this or that drug affects this or that issue, is it really a valid study? It’s as if they are saying, go out and do what you typical Americans do. eat donuts, processed foods and GMO everything and heaven forbid throw a moderate amount of exercise at the problem, because altering your lifestyle would not affect anything in the slightest, only drugs will resolve this issue. It’s a self fulfilling prophecy the drug companies perpetuate as they benefit most from any studies and any seeking a solution not related to drugs will rarely get the needed funding. It’s sad that this country has become so brainwashed and alternative view is vilified or ignored. Need I say more?
If you want to help your body absorb more Curcumin add pepper and Fulvic acid to the mix takes some time to work at least 3 months..
An easy to find fulvic acid product is shalijit it will also help with HBP and Diabetes
The “pepper” needs to be pepperine, not found in regular ground pepper but present in fresh ground pepper. Pepperine is volatile.
The flesh. And to think some think of a big bang theory.
I have two auto immune diseases. Grovers disease and PMR. I am constantly looking for ways to counteract these symptons. There is no cure for either one. For Grovers disease there is anti-itch lotions and for PMR I have been on 5mg prednisone for a few years. I tried tumeric in powder form for one month and
it did not help. Still looking for that magic solution. Someone mentioned Anatabine which I will research. Thanks.
I very much appreciate your article , being at that age where arthritis is a ‘reminder’ everyday. Luckily at this time I take NSAID’s occasionally, but I’m glad to see that Omega 3, which I take, is a benefit in reducing inflammation. Thank you for the article….. and the comments.
Doc, please investigate these links to various scientific studies on the use of curcumin for ra and other inflamatory diseases and injuries:
https://www.ncbi.nlm.nih.gov/pubmed/22407780
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3535097/
I’ve been using a curcumin preparation that is sold as Zyflamend for 4 years under the supervision of my MD for treatment of severe stenosis and peripheral nerve pain resulting from a ruptured lumbar disc. The curcumin compound enabled me to reduce ibuprofen from 1200 mg per day to 400 mg per day which made my doctor much happier.
Thanks for this. One area that has been “glossed over” in MY view is the LDL-C issue. At NO point is the issue of oxidised LDL v reduced/regular LDL addressed. This issue is critical to determining whether reducing LDL is necessary or whether reducing the oxidation of LDL is. It is apparently oxidised LDL that ends up in the cracks in the endothelium that are then covered in plaque which can cause cerebral or coronary events if they burst. What caused the endothelium to crack in the first place? We now know that the endothelium itself is the body’s largest organ and is responsible for producing Nitric Oxide which relaxes the arteries as well as having anti-inflammatory and anti-thrombotic capabilities until Endothelium Dysfunction sets in with the attendant disastrous health conditions.
I’d say the oxidation of LDL in the endothelium is the problem, not LDL by itself as it does serve other purposes. It only oxides in the presence of free radicals, and as you said it needs to find a way into the endothelium to begin with. Comes down to building and maintaining a strong immune system.
Quite the contrary. There is a significant amount of evidence that oxidation of ApoB-100 which is the protein that encases the lipids and cholesterol esters in LDLs results in prevention of the ApoB-100 receptor from assimilation of the LDL particles by tissues in proximity to the vessels.
As a consequence the oxidized LDLs are scavenged and collected by macrophages to such an extent that the macrophages appear foamy (creating foam cells). The macrophages become trapped creating plaque. When the plaque ruptures (labile plaque) platelets and clotting factors respond by forming a clot which occludes the vessel.
The native LDLs are meant to leak through the endothelium to enable surrounding tissues to assimilate the LDLs which contain cholesterol and lipids necessary for normal cell metabolism. The junctions between the endothelial cells are gap junctions to enable this leakage. I have not seen any description of cracks forming in the endothelium as initiating the problem, although I suppose that as the plaque enlarges it disrupts (cracks) the vessel leading to a clot.
There is evidence that it is not cholesterol per se but LDL particle concentration that increases risk of coronary artery disease. That is why it has been proposed that measuring ApoB-100 concentration is a better measure of risk. The confounding issue is that the use of statins which reduce cholesterol production reduce risk. It’s possible that with reduced cholesterol, lipids are packaged differently (LDL-C vs HDL -C) and the associated proteins in those particles are not as easily oxidized or alternately cleared by the liver quickly.
Measurement of ApoB-100 is complex and therefore uncommon and expensive.
I’ve asked my cardiologist if we could do this but he said my health insurance was not likely to pay for it. I was interested since I’m a retired biochemist and able to debate issues with my cardiologist.
There is a drug under development that prevents the ApoB-100 receptor from being degraded after it is assimilated into the cell with the LDL particle. With recycling of more receptor there is the belief more LDLs would be assimilated reducing serum concentration leading to less opportunity for oxidation and the whole cascade leading to a clot. This sounds good but it’s not clear to me how the increased cholesterol and lipids inside the cells would be metabolized.
With respect to all of the comments in this thread I believe there is no guarantee that any medicine or diet will prevent CAD. Perhaps antioxidants, a low fat diet and exercise help. It has been stated that a semi-starvation diet is the key to longevity. If we returned to the diet of humans about 150,000 years ago we would perhaps do better but then with the reduced lifespan of early man it’s possible that we would still suffer the same fate.
Nature only improves by experimentation through natural selection. Unfortunately that wouldn’t work either since these diseases take their toll with those of us in the selection shadow for humans (i.e. when we are no longer reproducing). Mutagenesis is the most powerful force affecting life. Catch 22.
Thanks for clarifying. Sounds to me like it is oxidation outside the endothelium that is the issue then. By what cause? Presence of free radicals alongside LDL & Apbo-100? Or simply too much LDL? Both? Neither?
I fixed my own minor celiac & diabetic symptoms by switching my eating habits to a mix of ketogenic, paleo and western price. Essentially a low or slow carb diet, high in good fats and protein. More important than any type of nutrient is how food is produced and processed. I’m fine with eggs from my own chickens for example, but I’ll throw up eggs raised on a high grain diet. I’m also fine with properly fermented sourdough bread, or fermented buckwheat pancakes, but not with any other foods containing grains or nuts. Some forms of processing are needed in order to remove certain anti-nutrients or toxins such as phytic acid. I’m slowly working on figuring out what the proper process is for each food. Oats for example, need to be ground into meal and and boiled before the anti-nutrients are neutralized, steel-cut or flaked is not good enough. For sweeteners I’m ok with molasses, maple syrup, honey and cocnut sugar, but not brown or white. When I first made the dietary changes I lost close to 20 pounds. I haven’t been sick since I switched 6 years ago, and as long as I avoid the sugars and grains/nuts/seeds the weight doesn’t come back.
My anti-inflammatory diet/supplements (low carb/no sugar/no grains) has healed me of a number of horrific — some thought to be incurable conditions. It all started when I decided I needed to deal with excruciating pain from a horrific ski accident. Our culture is so addicted to sugar in all its various forms we tend to dismiss it. I sure was. To your good health.
The first news was that statins achieve their effect by blocking cholesterol buildup. They do indeed block endogenous cholesterol synthesis by blocking mevalonate synthase, but not dietary. Then the news was that statins’ cholesterol effect is not the way they succeed. Inflammation is indeed likely the answer.
There have been some studies that show statins are only marginally effective at blocking coronary deaths. I suspect that once the cat is out of the bag, it is too late for statins to help. This Rosuvastatin study appears to be of individuals who don’t have cardiac disease. Too bad we can’t get statins before we get atherosclerosis!.
A shame that the result is some claim they are completely ineffective. Bad logic.
There was a recent Science News article on other effects of statins. Appears they are wonder drugs across the board.
True: some people react badly to them. Some people react badly to aspirin. Get over yourselves!
To consider: fat cells are inflammatory, as is glucose.
Very interesting article. I’ve been a follower for some time of Hyman. But I didn’t know he was a proponent of the bogus and debunked vaccine-autism-connection claim. This seems to be a bee in the bonnet of many of the “alternative” medicine world, although not all. I’ve heard more than one such doctor say forcefully that the claim is, in fact, bogus, which is the truth.
The evidence on the inflammatory and metabolic-syndrome-promoting SAD (standard American diet of highly processed foods) is pretty strong, as is the evidence against the 40-year-long public health disaster of the “diet-heart” hypothesis, that eating saturated fat is bad for you. It isn’t; quite to contrary. From my own clear personal experience with moving away from a carbohydrate-rich diet toward eating more saturated fat and high-quality animal protein, my own blood lipids have improved a lot, especially triglycerides. Even my once chronically low HDL is now higher.
Quote from Michael. Fossel’s Book Telomerase Revolution:
circulating monocytes and platelets begin to adhere to the endothelial wall, even before the lower layers show changes. But as endothelial failure progresses, the result is increasing inflammation in the subendothelial layers, as macrophages and other immune system cells begin to enter the arterial walls. Scarring follows soon after, and cholesterol begins to accumulate on the scar tissue.
3D Signatures Inc OTCQB: $TDSGF np
DXD – http://www.3dsignatures.com/investors/
I looked up some articles by Dr. Hyman, and he doesn’t claim that vaccines cause autism flat out. This article discusses his experience with a young boy suffering from severe autism and simultaneously many somatic problems:
http://drhyman.com/blog/2010/05/19/why-current-thinking-about-autism-is-completely-wrong/
The real gist of it has to do with gut inflammation (leaky gut, gluten, poor nutrient absorption, chronic failure to expel toxins, and much more). And of course, we now know that this connection (brain and gut) is real and powerful, not at all folklore. Vaccines come in secondarily, as the possible cause of a strong allergic reaction. It’s not the main point Hyman is making.
Steps ..baby steps as Autism Researchers Discover Genetic ‘Rosetta Stone’ #science https://www.ucsf.edu/news/2017/01/405631/autism-researchers-discover-genetic-rosetta-stone
via https://twitter.com/drnic1/status/828049508890185731