Aside from the tiny minority of people who can actually say what each of those initials stand for, we can divide the populace into two groups – those who specifically know that CRISPR is a tool used in gene editing and those who know (or figure) that it’s a highly promising but exceedingly complex technique for addressing difficult-to-treat medical conditions or diseases. And of course there are those who conclude that CRISPR must be something that makes things more crispy, like putting soggy French fries in the oven.
We’ll get to spelling out the acronym a bit further on in this piece – and believe me, just spelling it out won’t make you understand the way it works. I will attempt a clear explanation, not only of the acronym, but of how CRISPR works in gene editing. But in the meantime, a bit of background on the larger subject of gene editing itself.
Gene manipulation in some form has been around for a long, long time – millennia, dating from those times when nobody had the foggiest notion of what a gene was, or how it was that some but not all of the characteristics of the parent were passed on to the offspring. The term “parent” as used here does not refer to a human Daddy or Mommy but to whichever being, animal or vegetable, that gave rise to offspring. When hunter-gatherers selected the largest fruit from trees, brought these prizes back to their huts for dinner, and threw the pits on the dung heap, they were unknowingly practicing genetic manipulation. From those seeds the next generation of fruit trees would grow, producing bigger fruit than their cousins in the woods. Tulip fanatics in Holland in the 17th century were doing genetic manipulation, as were breeders of prized hogs, as well as the agronomists who gave us such mixed-breed fruits as the pluot (a cross between a plum and an apricot) and perhaps the grapefruit (a cross between a sweet orange and a pomelo, which looks like a grapefruit but is quite a bit larger, and more sour). The grapefruit may have emerged accidentally, or it might have been a deliberate creation.
In any case, what the creators of those hybrids (and many, many more) were doing was taking genetic material from different “parents” and putting them together such that the offspring are genetically modified – essentially different from their parents. But they had no idea that all characteristic features of the animals and plants they were working with were encoded in genes, nor yet even of the existence of genes.
The concept of genes and genetics came into being in the early years of the 20th century, although as yet nobody knew what a gene actually was. Chromosomes had been identified as related to inherited characteristics, and had even been observed under the microscope at that time, but what the chromosomes actually were was still unknown. As the capacities of microscopes progressed, it became possible to get a closer look at chromosomes, which led to the discovery in 1953 by Francis Crick and James Watson of the configuration of the DNA molecule in the chromosome, i.e., the famous double helix. DNA in turn was found to be composed of a linking of four amino acids, termed “bases” (adenine, cytosine, guanine and thymine plus a phosphate group and a pentose sugar). The human genome consists of about 6.2 billion bases, paired and linked together in a very long twisting chain, which is wound around itself so as to take up minimum space. The genetic information is carried by these 3.1 billion base pairs, and the possibility of effecting changes to that genetic information, such as eliminating the genes that carried the information that would cause diseases or disabilities, was immediately the subject of research.
The information carried in the genome might be compared with the information embodied in computer code. As by now everyone knows, computer code as read by the computer consists of strings of ones and zeros, which signify yesses and nos – i.e., an impulse transmitted or an impulse blocked. The human entering the code does not have to translate the numbers in our decimal system into the binary system of ones and zeros – the computer takes care of that. There doesn’t seem to be any limit to the amount of information that computer code can convey, but those strings of ones and zeros can get pretty long.
Genetic information is conveyed by that string of amino acids. As in computer code, the information is essentially digital, except that instead of two digits to work with, the genome can use four digits, for the four amino acids – A, C, G, and T. Thus, it can carry a good deal more information than computer code, in a limited space.
Tinkering with the genome in a way that would provide health benefits became a goal of scientific and medical research. One could call it the “holy grail.” But it was far from simple. A couple of early attempts to do actual gene editing were exceedingly complicated and difficult. One method relied on what were termed “zinc finger nucleases” (ZFNs). These would have to be designed specifically to recognize and bind to specific fairly short base-pair sequences, and sometimes editing a single short sequence would require the design and construction of three ZFN domains; longer base-pair sequences would require more elaborate ZFN domains.
The finding that opened the path to what we now call gene editing was the discovery that in those long strands of DNA, the gene sequences were separated by clusters of sequences that did not appear, at first glance, to convey any genetic information at all. We might call them nonsense sequences. They followed a certain pattern – they read the same way from either end: i.e., they were palindromic. They occurred at repeated intervals in the DNA strand. Thus, they came to be called Clustered Regularly Interspaced Palindromic Repeats – “CRISPR”. These sequences were discovered in 1980 and were originally dismissed as being of no interest – “junk DNA.”
It turned out that the CRISPR sequences, along with a CRISPR-associated (CAS) enzyme are of vital importance in conferring immunity to repeated infection by a virus. This was discovered in research on bacteria, which also (of course!) have CRISPR in their genome. When a virus invades a bacterium, the bacterial CAS enzyme snips off pieces of the viral genome, and stores it in the CRISPR part of the bacterial genome so that the bacterium essentially remembers the virus that attacked it and can mount a defense. A number of these enzymes have been discovered, but the one – CRISPR-Cas9 – that has up to this point been found to be most useful in gene editing was discovered in 2012 by Jennifer Doudna, at that time at the Broad Institute (Harvard / MIT), and Emmanuelle Charpentier of the Max Planck Institute for Infection Biology in Berlin.
In 2020, Charpentier and Doudna received the Nobel Prize in Chemistry in recognition of their work. The Chair of the Nobel Committee for Chemistry, Claes Gustafson, said “There is enormous power in this genetic tool, which affects us all. It has not only revolutionized basic science, but also resulted in innovative crops and will lead to ground-breaking new medical treatments.”
The Nobel Committee’s statement went on as follows:
“Since Charpentier and Doudna discovered the CRISPR/Cas9 genetic scissors in 2012 their use has exploded. This tool has contributed to many important discoveries in basic research, and plant researchers have been able to develop crops that withstand mould, pests and drought. In medicine, clinical trials of new cancer therapies are underway, and the dream of being able to cure inherited diseases is about to come true. These genetic scissors have taken the life sciences into a new epoch and, in many ways, are bringing the greatest benefit to humankind.”
CRISPR-Cas9 has been central in gene editing for the past nine years. The Cas9 enzyme makes it possible to “snip” entire gene sequences out of the genome without altering the gene itself. The Cas9 enzyme is guided by RNA, which has the capacity to locate specific DNA sequences. The enzyme binds to and breaks the CRISPR cluster on each end of the genetic material in a way that permits the genome to link together without a permanent break, resulting in the deletion, or “knock out”, of that gene. The word “snip” is of course used figuratively; there is no mechanical instrument that cuts the CRISPR cluster. The binding action of the enzyme separates the individual particles in the cluster. And the binding action is possible because of the configuration of the enzyme and the electric potential of its components.
The figure below was created by the Nobel Committee to attempt to explain how CRISPR works:
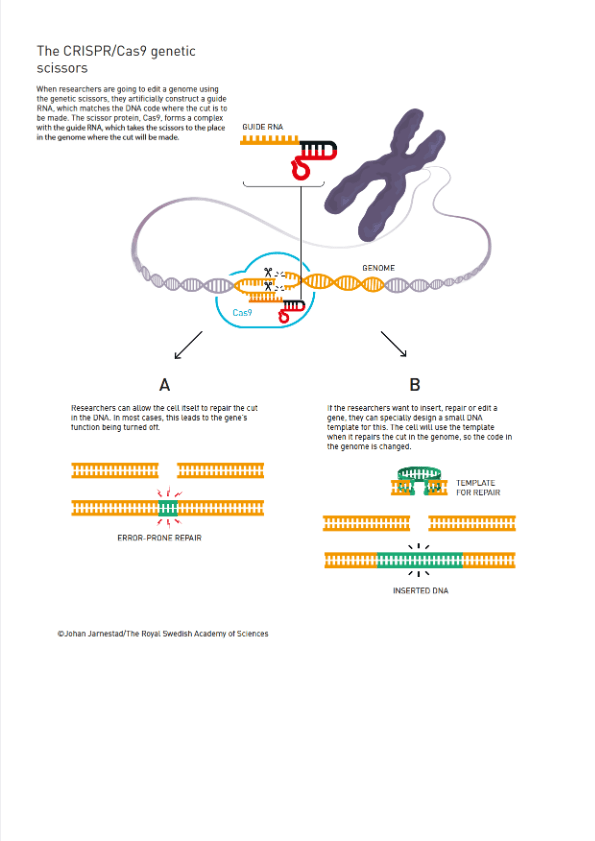
Using gene editing to prevent some inherited diseases
The dream would be to cut out the genes that doom people to a number of diseases, such as amyotrophic lateral sclerosis (ALS), sometimes known as Lou Gehrig’s disease. A person one of whose parents has the ALS gene has about a 50% chance of developing the disease, which reduces the life expectancy of the patient to less than five years after onset of symptoms. In about 10% of ALS cases, the cause is a genetic factor. And a number of other diseases have a major genetic component. For example, cystic fibrosis, Down syndrome, Duchene muscular dystrophy, hemophilia, Tay-Sachs disease, Tourette syndrome, and many others.
Genetic manipulation in the interest of protecting people from disease may not be as clearly a good thing as we might assume at first glance. For example, take sickle cell anemia, an inherited genetic disease, which is the fourth leading cause of death in children in many less-developed countries. Sickle cell presents a contradiction. One would expect natural selection to weed out a gene that has such dire consequences, but that isn’t happening. About 4.5 million people currently have the disease, and another 43 million have the sickle cell trait, but have not shown signs of the disease. A possible reason for this is that persons having the sickle cell trait – a single gene – have an approximately 30% lower probability of developing malaria, which is a major killer in the parts of the world where sickle cell is prevalent. So if scientists were able to eliminate the sickle cell gene, is it not likely that the number of malaria cases – and deaths – would zoom?
A company co-founded in 2013 by Emmanuelle Charpentier and Jennifer Doudna, entitled CRISPR Therapeutics, has developed (jointly with Vertex Pharmaceuticals) a drug, CTX001 for the treatment of sickle cell disease and Beta thalassemia, a rare blood disorder. CTX001 received orphan drug designation from the FDA in May of 2020. Charpentier and Doudna are well aware of the link between sickle cell disease and malaria, and have taken precautions that tinkering with the sickle-cell trait would not decrease the likelihood that the gene would continue to provide protection from malaria. CRISPR Therapeutics gave rise to another outfit called Casebia Therapeutics, which came under the control of CRISPR Therapeutics in 2019 and is moving forward as a joint venture with Pfizer.
Clearly, tinkering with the human genome is not an area that should be entered blithely. There was a great deal of public controversy after the announcement, a few years ago, that a scientist in China had performed gene editing on a human embryo such that the babies, twin girls, were born with an edited gene that was supposedly associated with a much lower risk of HIV. Opinion on the question of actually altering the human genome was starkly divided. One faction asserted that it must never be done under any circumstances, pointing to the possibility of errors that would then be perpetuated in the offspring of those “designer babies.” Another possibility that was met with much suspicion and antagonism was that these “designer babies” would actually be superior beings – physically, intellectually, or in some other way – and grow up to lord it over the rest of us. But a contrary argument was that if it were indeed possible to edit out the genes that greatly increased the risk for a number of disabling or deadly diseases, enacting laws that deprived humans of that possibility might be considered equivalent to outlawing vaccinations. The possibility of error, however, hovers over the entire field of gene editing. How do we know, for example, that cutting out the gene that is associated with an increased likelihood of Down syndrome does not also have another, perhaps even more unfortunate result?
An area which doesn’t attract much disapproval on ethical grounds is manipulating the genes of mice. For more than 30 years, genetically-altered mice have been used in scientific research. It’s very handy to have a supply of mice whose genes have been altered in such a way as to make them especially likely to develop the diseases that are being targeted in humans. In any number of studies of important drugs, the candidate drug is evaluated in transgenic mice prior to any kind of study in humans.
"reveal" emails? If not,
just click here...
Initially, efforts to manipulate the genes of mice employed chemicals or viruses. These methods were clumsy, and resulted more often than not in random mutations that were of little clinical use. Genuine gene editing, first using the above-mentioned zinc finger nucleases (ZFNs) and transcription activator-like effector (TALE) nucleases, and later using the CRISPR-Cas9 system, have resulted in much more efficient and specific gene editing. So now, researchers can work on diabetic mice, mice with heart disease, cancer, and numerous other medical conditions. They are by no means identical to humans in terms of their response to drugs, but they are hugely helpful.
Gene editing and our own immune response
One of the most promising areas for the application of gene editing is our own immune response. As you have read in previous Doc Gumshoe screeds, especially about COVID-19, there is a constant war raging in our bodies between our immune system and harmful invaders of all kinds, whether these are microbes, viruses, foreign substances that don’t belong inside our bodies, and also our own cells when they mess up their own reproductive processes and become cancerous. The chief patrolling agents are T-cells, which are highly effective against a wide range of enemy agents, which they attack and kill. T-cells are equipped with receptors on their surface that permit them to recognize our own healthy cells and instruct the T-cells to leave those cells alone. These surface receptors, called immune checkpoints, act as brakes, so that the T-cells limit their attacks to the dangerous invaders. But cancer cells, with their own survival at stake, have developed high levels of proteins that also attach to those checkpoints, essentially fooling the T-cells into mistaking some cancer cells for normal cells, which then sneak through the protection of our immune response.
Those proteins have been labeled “checkpoint inhibitors,” and they were an important target of drug development for 25 years before the approval of the first drug that addressed the checkpoints on T-cells in such a way as to get around those survival mechanisms in cancer cells. That was ipilimumab (Yervoy), from Bristol-Myers Squibb, which received FDA approval in March 2011 for the treatment of malignant melanoma. Several other drugs followed, including some of the most successful (and hugely profitable) cancer drugs, including nivolumab (Opdivo, also from BMS), and pembolizumab (Keytruda, from Merck).
An early gene-editing success
Gene editing has been successfully applied to T-cells in such a way as to direct them to attack cancer cells, overcoming the defense mechanisms that cancer cells mount to evade treatment. One of the first patients to be treated in this way was a seven-year-old girl who had relapsed acute lymphoblastic leukemia (ALL). The majority of children with ALL achieve complete remission after about two years of chemotherapy, but Emily Whitehead was in that 15% who were resistant to even the most intensive chemotherapy.
She was initially treated with chemotherapy, and when that first round of treatment failed, her parents took her to the Cancer Center of the Children’s Hospital of Philadelphia (CHOP), where the oncologist recommended a second round of chemotherapy for Emily. When she failed that second round, in February 2012, her parents agreed to enroll Emily in a clinical trial of advanced experimental T-cell therapy for advanced B-cell leukemias and lymphomas.
The therapy consisted of collecting Emily’s T-cells and modifying them genetically to attack the leukemic cells, termed B-cells. Emily’s T-cells were harvested and treated with an agent that incorporates a gene that encodes for a specific antigen receptor that recognizes and attaches to a protein called CD19 that is found only on the surface of those B-cells. The engineered T-cells were then reintroduced into Emily’s system, where they spread throughout her body and began to find and kill the cancerous B-cells. Genetically engineered T-cells are known as chimeric antigen-receptor modified T-cells, thus CAR-T and CRISPR CAR-T, since the CRISPR sequences are what permit the T-cells DNA to be edited.
Emily’s parents had been warned that the treatment was not without risk. They had expected that Emily would experience flu-like symptoms, but what took place was a good deal more serious. She was admitted to pediatric intensive care where it was learned that the growing presence of T-cells in her body gave rise to the proliferation of a cytokine that is involved in rheumatoid arthritis. The cytokine, interleukin-6 (IL-6), is customarily treated in patients with rheumatoid arthritis by tocilizumab (Actemra, from Genentech). Emily was treated with tocilizumab and her condition improved markedly. Almost overnight, her breathing improved, her fever dropped, and her blood pressure was back to normal. In the ensuing weeks, Emily complete recovered from her adverse reaction to the T-cell infusion, which is called cytokine release syndrome.
The doctors who were supervising her treatment were still unsure whether her underlying ALL was being successfully addressed by the T-cell treatment. Several weeks after the initial infusion, a bone marrow test was performed to assess the results. The medical team concluded that Emily was in complete remission. The bone marrow test was repeated after another three-month interval, and then after six months, and she had no signs of the disease. At the present time, seven years after the T-cell infusion, she continues to be entirely disease-free, and the descendants of the original genetically-modified T-cells are still circulating in her body, armed and ready to attack any signs of cancer.
About six years ago, Emily Whitehead’s family started the Emily Whitehead Foundation, which this past January funded the Children’s Hospital of Philadelphia’s study of the neuropsychological outcomes of pediatric CAR-T-Cell therapy. Their mission statement is:
“Our mission is to raise funding for CAR T-cell therapy research because this treatment is less toxic and more targeted than standard chemotherapy or radiation. Less toxic treatments for treating childhood cancer are greatly needed because children have to live with the harsh side effects of standard treatments for much longer than adults.”
CRISPR CAR-T treatment targeting the CD19 protein has been used in several hundred patients with relapsed or refractory ALL, with mostly positive results; however, research continues on the best treatment modalities for this disease. A strategy that is under consideration for patients with factors that put them at high risk for refractory ALL is to harvest a supply of T-cells in advance, so as to have them ready for gene editing and reinfusion if the need arises.
Applying CRISPR gene editing to drug development
I should point out that in the CRISPR world, the drugs are not directly given to the patients. What happens is that the patient’s T-cells are harvested and exposed to the chimeric antigen receptor (CAR), which is derived from mice. In Emily’s case, this receptor specifically targeted the CD19 protein, which is what made the T-cells able to find, attack, and destroy the cancer-causing cells. Then, the T-cells are reinfused into the patient’s body where they multiply and perfuse the patient’s circulatory system. The medical team then crosses its fingers and waits for the edited T-cells to do their work, which they mostly do.
At this point, seven drugs developed through gene editing have been approved by the FDA. The first was Kymriah (tisagenlecleucel, from Novartis), which is approved for the treatment of relapsed/refractory B-cell precursor acute lymphoblastic leukemia, the form of cancer that Emily had. The second was Yescarta (axicabtagene ciloleucel, from Kite Pharmaceuticals, which is approved for the treatment of relapsed/refractory diffuse large B-cell lymphoma.
As I write this, the news was released that on Friday 26 March the FDA approved another CAR-T derived drug, idecabtagene vicleucel (ide-cel, Abecma) from Celgene, part of Bristol-Myers Squibb. It is the first cell-based gene therapy for multiple myeloma, currently a disease with no known cure. The new drug is approved for patients whose disease progressed or did not respond to four previous lines of therapy, including an immunomodulator, a proteasome inhibitor, and an anti-CD38 antibody.
Primary supporting data for the approval came from a Phase 2 trial involving 127 patients with relapsed/refractory myeloma, 100 of whom could be evaluated for response. The results showed an overall response rate of 72% and stringent complete responses (sCR) in 28% of patients. The median time to response was 30 days, and the median duration of response was 11 months, increasing to 19 months for patients who achieved sCR. Stringent complete response in multiple myeloma is a higher standard of response than complete response.
Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in the agency’s announcement on Friday. “While there is no cure for multiple myeloma, the long-term outlook can vary based on the individual’s age and the stage of the condition at the time of diagnosis. Today’s approval provides a new treatment option for patients who have this uncommon type of cancer.”
Another drug recently approved by the FDA is Breyanzi (lisocabtagene maraleucel, from Juno Therapeutics in collaboration with Celgene). It was given the thumbs up on 5 February of this year for the treatment of adult patients with relapsed or refractory large B-cell lymphoma who had experienced two or more lines of systemic therapy.
Breyanzi is a CD19-directed chimeric antigen receptor (CAR) T-cell immunotherapy. It consists of autologous T cells that are genetically modified to produce a CAR protein, allowing the T cells to identify and eliminate CD19-expressing normal and malignant cells. This is the same mechanism used in Emily’s treatment, prior to the development of the drug.
The efficacy of Breyanzi was evaluated in TRANSCEND, a single-arm, open label, multicenter trial in adults with relapsed or refractory large B-cell lymphoma. Of the 192 patients evaluable for response, the overall response rate was 73%, with a complete response rate of 54%. The median time to first response was one month. Of the 104 patients who achieved complete response, 65% had remission lasting at least 6 months and 62% had remission lasting at least 9 months.
Other gene-editing-derived drugs are Imlygic (talimogene laherparepvec), from BioVex, a subsidiary of Amgen; also Provenge (sipuleucel-T), from Dendreon; Yescarta (axicabtagene ciloleucel) from Kite Pharma; and Zolgensma (onasemnogene abeparvovec-xioi), from AveXis, Inc.
You might have noticed that several of these drugs have the suffix “leucel” attached. This signifies that they are associated with lymphocytes, monocytes, or antigen-presenting cells. Similarly, the “vec” ending refers to the vector that is employed to deliver the agent. Those long complicated names weren’t pulled out of a hatful of Scrabble tiles. They actually say something about the composition of the drug.
The success rate for CAR-T therapy is a bit equivocal. The initial remission rates look excellent, sometimes running to 90% or higher. However, long term survival is another matter. For example, in the case of therapies that target the CD19 protein, not all the leukemic cells express CD19 – a few express other cytokines, and after the CAR-T therapy has eliminated those B-cells with the CD19 protein, other B-cells start multiplying and spreading.
To attempt to combat this, research is ongoing on the possibility that T-cells could be marked with other antigens, such as the BCMA protein, which is commonly expressed in multiple myeloma, or other antigens such as CD20. There are also efforts underway to engineer CARs targeting many other blood cancer antigens, including CD30 in refractory Hodgkin’s lymphoma; CD33, CD123, and FLT3 in acute myeloid leukemia; and BCMA in multiple myeloma.
The merits of “staying alive”
It’s obvious that treatment options based on gene editing would be reserved for patients who have essentially run out of options. By the time they are considered for these treatments, they are very ill and soon facing death. CRISPR CAR-T or related forms of therapy will give these patients perhaps as much as a year of additional life. But the cost of such therapy is high, not only in economic terms, but also in terms of side effects. The question inevitably arises, “it it worth it?”
In baseball, “staying alive” describes what the batter is doing when he fouls off pitch after pitch, waiting and hoping for the pitch that he can swat for extra bases. The process is a bit boring for the fans, exhausting for the pitcher, and more than a bit vexing for the opposing team. But sometimes staying alive pays off.
What happens sometimes is that, in those additional months of life that were granted to the patient by means of these treatments, a new and more effective treatment option becomes available. That’s one of the great benefits of staying alive.
As we all know, regardless of the effectiveness of medical treatments, they can do no more than postpone our eventual end. But even a few additional years of reasonably sound health can be a priceless gift, and that is what treatment based on gene editing is seeking to deliver. I strongly agree with the words that accompanied the Nobel Prize to Charpentier and Doudna _ “These genetic scissors have taken the life sciences into a new epoch and, in many ways, are bringing the greatest benefit to humankind.”
* * * * * * *
Doc Gumshoe has done no more than a surface dive into gene editing – not by any means a “deep dive.” Although it’s the best known technique of gene editing, CRISPR Cas9 is by no means the whole story. A couple of researchers at the Broad Institute are using a different protein that targets RNA rather than DNA. The protein, labeled Cas13, permits CRISPR to recognize and target infectious diseases that use RNA as their genetic material.
The Cas13 protein is employed in a new system that may one day be used both to diagnose and treat a large number of infections, caused both by known and by emerging viruses. The system has been called CARVER for Cas13-Assisted Restriction of Viral Expression and Readout.
Another gene editing tool saw the light of day just a couple of years ago. This one has been called PRIME. The chief difference between PRIME and CRISPR-based methods is that while CRISPR relies on the ability of cells to divide to help make the desired changes in DNA, PRIME does not require that trait to function. That means that PRIME could be used to correct genetic mutations in cells that do not commonly divide, such as those in the nervous system. Many diseases, such as Parkinson’s and Huntington’s, are caused by mutations in nervous system cells.
To give you an idea of the volume of interest in these areas, gene editing is the subject of more than 200,000 published papers, and PRIME alone is being studied in 1,808 clinical trials. So there will be quite a lot more to report on. I will try to keep my head above the flood and share what I learn with Gumshoe Nation.
Thanks and best to all, Michael Jorrin (aka Doc Gumshoe)
[ed. note: Michael Jorrin is a longtime medical writer (not a doctor), who I dubbed “Doc Gumshoe” many years ago — he writes health and medicine-focused columns for our readers a couple times a month, and though he does not generally cover investment ideas he has agreed to our trading restrictions. You can find his past columns here.]
What’s your take on RLFTF’s Zyesami?
Thanks.
I had never heard of it, and when I checked there was not a whole lot of information. What I did turn up was encouraging – that is, the results of the Phase 2B/3 trial in patients with COVID-19. I cannot comment on the stock as an investment. That’s not my department.
Thanks!
Interesting and informative article on CRISPR development!
Great story, have lots of Edit and Crisper in my portfolio.
Do your next write on the Proteomics Space and SEER
As far as yielding any benefit in terms of treament, proteomics is in its infancy. The number of proteins is colossal. The research will surely turn up a great deal of interesting stuff, but there’s not a whole lot for me to report on at this point.
If my understanding of the transcript of the last earning call for SEER is correct, Seer has gone from setting our initial vision of enabling broad scale access to deep unbiased proteomics to shipping products to our customers in just three short years. We have built a world-class team that is second to none in the proteomics space and we have a product solution that is well-positioned to enable the kind of proteomic studies that were just not possible before Seer.
You can pull the report from their web site.
Now, in order to access a broad range of proteins and especially those that are of low abundance, researchers are forced to follow laborious, expensive, depletion and fractionation methods to methodically separate proteins before detection. These methods simply do not scale well, are accessible to few labs and are cumbersome to perform. Our Proteograph Product Suite replaces these methods with a streamlined, automated, workflow that can fit in front of nearly any detector delivering unmatched access to proteomics information. With the Proteograph Product Suite, we have reimagined the entire approach to unbiased proteomics, delivering a solution that can enable nearly any lab to adopt large scale proteomics and notion that was previously inconceivable. Now our Proteograph Product Suite is detector agnostic, providing the solution that makes access to large scale deep unbiased proteomics tangible and importantly, not just for a select group of thought leaders in proteomics but for really any lab wanting to do unbiased proteomics studies.
You are amazing – sounds like you have a PhD in medicine!!
Another excellent article by Doc Gumshoe to explain a complicated medical topic to we average readers in terms & language we can understand. As a small investor, I’m not going to chase or invest in the companies involved in this cutting-edge research as that’s beyond my risk level.
I do urge Doc Gumshoe to consider tendering his articles to a company that does Biology textbooks for high school and college level students with the view to getting royalties.
You write, “DNA in turn was found to be composed of a linking of four amino acids, termed “bases” (adenine, cytosine, guanine and thymine plus a phosphate group and a pentose sugar).” Oops. Proteins are made from amino acids. DNA is made from nucleotides, of which there are four types with those four bases, A, C, G and T.
Thanks for the fix, best, MJ
You seem to have ignored Sangamo’s Zinc Fingers technology that they have been working on for over 20 years and have 17 projects at various stages of development. They too are partnering with the largest pharmaceutical companies.
I have a question about COVID-19 and immunity and something Doc Gumshoe wrote goes with it. He wrote in the article above: “It turned out that the CRISPR sequences, along with a CRISPR-associated (CAS) enzyme are of vital importance in conferring immunity to repeated infection by a virus. This was discovered in research on bacteria, which also (of course!) have CRISPR in their genome. When a virus invades a bacterium, the bacterial CAS enzyme snips off pieces of the viral genome, and stores it in the CRISPR part of the bacterial genome so that the bacterium essentially remembers the virus that attacked it and can mount a defense. ”
So, if this is the case, as I understand it, then when I get COVID-19 my body should should snip off the viral genome and store it in the CRISPR part of my genome. Thus, I should build the immunity to COVID-19. However, regarding COVID-19, confusion reigns and I’m seeing mixed reports. Since I had it last November am I immune or not? Is it time sensitive, i.e. I’m immune for a time but not forever? If so, why is the vaccine different since I assume the vaccine does what getting the disease does without me needing to get sick.
Am I totally misunderstanding this?
If not, any thoughts/answers on this?
Thanks Doc Gumshoe!
Can CRISPER be used to reverse Parkinson’s ?
Not yet. A potential target might be the APOE3 gene. Don’t hold your breath.
Travis: I appreciate the enormous amount of information presented in this piece and time required for Michael Jorrin to do the necessary research to write this article. But there are glaring fundamental errors of biochemistry and molecular biology that required editing by a molecular biologist or a cell biologist and correction prior to publication on your web site: The mistakes mar an otherwise very useful and interesting article and undermines his credibility to present future information on the topic covered.
Nucleotide bases are not amino acids. Please! The latter are linked together in long polypeptide chains that form proteins. The Central Dogma of biology is that the genetic information of DNA is copied into messenger RNA (m-RNA) and translated by ribosomes in the cytoplasm of the cell using Transfer RNA (t-RNA) molecules linked to Amino acids to form polypeptide chains that fold spontaneously to form 3D structures called proteins. It is the unique folding of AA’s into 3D structures that enable proteins to carry out enzymatic and structural roles in biological organisms.
The 2D sequence of Nucleic acids in DNA and RNA molecules code for the sequence of amino acids that form proteins. The coding is based on triplet sequences of nucleotide bases adenine (A), cytosine (C), guanine (G), thymine (T). Each nucleotide base is covalently bonded to a deoxyribose sugar-phosphate molecule which are all alike and in turn bond to each other to form the backbone of long chains of nuclei acids of a strand of a DNA molecule. A DNA molecule is composed of two separate strands or chains of nucleotides (+ and -) that bind to each other to form the famous double helix.
To use an analogy of a ladder, chains of deoxyribose sugar-phosphate molecules link or bond together to form the stable backbone of the separate strands of the DNA double helix (the sides of the ladder), the complementary nucleotide bases (A, C,G, and T)of the separate strands form the rungs that are linked by H bonds.
The nucleic acid bases of the separate strands are complementary and bind by Hydrogen bonds across the strands such that A binds with T and G binds with C to form the rungs of the ladder of the DNA helix. So the H bonds between complementary nucleotide bases on separate strands of DNA is what keep the DNA molecule together and remain stable and replicable for decades during a person’s lifespan.
It is the unique sequence of nucleotide bases (A,C,G,T’s) that is the information content of a DNA molecule, particularly the relationship between nucleotide triplets or codons to specific amino acids: so the codon AGT codes for a different amino acid than say ACC or CAG. A protein may be composed of hundreds or thousands of amino acids hence the genes that rise to proteins are composed of sequence of bases more than 3x the number of amino acids.
Since there are 4 nucleotide bases, there are 4x4x4 or 64 possible combinations of triplet nucleotide bases. There are about 20 amino acids so there is redundancy in the codons especially for the third letter of the triplet, which is referred to as degeneracy, such that AGT and AGC or AGG could Code for the same amino acid. Other codons carry other information ie tell the ribosome where to start translation of the message and where to stop.
The coding relationship between triplets of nucleotide bases or codons and specific amino acids is referred to as The Genetic Code and required years of basic research to figure out, and followed the molecular structural break through of Watson and Crick in 1953 who proposed the double helix structure of DNA, with a major assist from the X-ray crystallography of DNA provided by fellow scientist Rosalind Franklin, who never got the credit she deserved for the value she provided to Crick and Watson.
It is of also of historical and scientific interest that a different group of Molecular Scientists figured out the Genetic Code rather than Watson or Crick.
The complementary nature of the base pairs on the separate strands of DNA in the double helical structure suggested how the replication of DNA to make exact copies of itself during cell division might occur. Such that the sequence of bases on one strand could give rise to its complementary strand by nuclear enzymes referred to as DNA polymerase. different cellular functions of short RNA chains also include controlling or inhibiting the expression of m-RNA of particular genes lending itself to different strategies of genetic therapy.
RNA differs from DNA by the substitution of a Ribose sugar in place of Deoxyribose, and the base Uracil for Thymidine, as well as its single stranded composition & structure. The covid-19 Virus is a RNA virus, a short single strand or chain of nucleic acids that comprise about 30 genes that code for its spike and capsid proteins and cell receptors.
Whereas DNA is structured for its replicable stability and long term preservation of genetic information, not only for a person’s lifespan but for future generations, RNA molecules do a wide variety of cell functions such as carrying the genetic message (m-RNA) from the DNA in the cell nucleus to the cytoplasm, the reading of the message as part of the wonderfully complex protein structures or Gene Machines called Ribosomes (ribosomal RNA) and the transfer molecular agents (Transfer-RNA or t-RNA) that are covalently linked to individual amino acids encoded for by the triplet codons read in sequence by the ribosomes. Different parts of the ribosome read the m-RNA codons in sequence and act as docking stations for the right t-RNA linked to the appropriate AA which are then joined together by peptide bonds to form the polypeptide chain of the nascent protein. Dozens of ribosomes are simultaneously reading a single m-RNA message . Quite a wonder.
BTW: Down’s syndrome is not caused by a single gene mutation but rather an extra chromosome, the non disjunction of Chromosome 21 during meiosis in formation of ova prior to fertilization. .So the Down’s Syndrome child has an extra copy of Chr 21.
Sickle cell disease and cystic fibrosis are due to single nucleic acid base mutations in the genes that give rise to alterations in Hemoglobin in Red Blood Cells and the Sodium ion pump or Na ion membrane transport protein in epithelial cells that line the respiratory tract and other organs. The Adult Hemoglobin molecule is comprised of 2 alpha and 2 beta chains. Sickle cell is caused by a single nucleic acid base change in the Beta chain. In 1957, V.M. Ingram of Cambridge University showed that sickle Hemoglobin differs from normal hemoglobin by the change of a single amino acid in the Beta chain; at position 6, glutamic acid is replaced by valine. The point mutation causing sickle cell disease if both parental chromosome copies of Hemoglobin B gene are mutated (ie both parents have sickle cell trait). The single gene, single base mutations of Sickle Cell disease and Cystic Fibrosis lend themselves far more readily to CRISPR editing than Down’s Syndrome and the former more so than the latter since the genetic effects are restricted to the membranes of RBC’s in the bone marrow in sickle cell disease whereas the mutation of CF gene simultaneously affects many epithelial surfaces in multiple organs.
References:
1 The Double Helix. A Personal Account of the Discovery of the Structure of DNA. James D Watson. 1968.
2. Molecular Biology of the Gene. James D Watson, Nancy H Hopkins, Jeffrey W Roberts, Joan A Steitz, and Alan Weiner. 4th Edition, 19$9.
3. Gene Machine. The race to discover the secrets of the ribosome. Venki Ramakrishnan. 2018.
Gerard, many thanks for your comment. I have not yet digested the content, but I will save it and read it carefully.
hi is there some thing serious about GSHD 13% –
If you’re talking about Goosehead Insurance, that’s outside my range.
I’m not aware of any particular GSHD news this week. It’s a very volatile and relatively small stock, so it does tend to make big moves if there’s any flow of investors in or out — negative news coverage, a newsletter selling or buying, etc. , but I don’t have a guess at the cause of the most recent dip.
Sir, The article gives very useful information about CRISPR technology for gene editing. I have couple of questions. What is the difference between stem cell therapy and therapies using CRISPR ?
I have read somewhere in this website about using CRISPR to treat alzheimer type of diseases. But your article indicates CRISPR cannot be used for repair of cells in brain. could you please explain.
Sorry, but I cannot answer this just now – we’re leaving in the morning for a short vacation. Your question will be answered in the next Doc Gumshoe piece. Thanks & best!
I would also like know the difference between stem cell therapy and gene editing.
Thanks for this information. Could provide a bit more detail about the vector used?
Are any of the RNA vaccines related to any of this?
Worrying development for CRISPR stocks. A recent study has flagged a new safety signal that could potentially hurt the drug developers focused on CRISPR–Cas9 gene editing. The condition known as chromothripsis has the potential to cause cancer eventually, according to the study : https://seekingalpha.com/news/3730324-edit-stock-is-lower-as-new-study-sheds-light-on-new-safety-concern-in-crispr-gene-editing.
This condition does not apply to Beam Therapeutics, which uses a base editing technique, but its not necessarily positive news for Beam either (which I own) in the short-term as the whole sector tends to move in tandem.